e-Publications
Boston ACS Meeting 2002 |
back to e-Publications
|
EXTRACT FROM CHEMICAL INFORMATION AND COMPUTATION 2002
|
||||||||||||||||||||||
| |
||||||||||||||||||||||
INTRODUCTION
|
||||||||||||||||||||||
| |
||||||||||||||||||||||
NEWS, NEW PRODUCTS AND THE EXHIBITION
|
||||||||||||||||||||||
| |
||||||||||||||||||||||
CINF AND COMP TECHNICAL PROGRAMMES
|
||||||||||||||||||||||
| Position: | n1 | n2 | n3 | n4 | n5 | n6 | n7 | n8 | n9 |
| Key: | 2 | 3 | 5 | 2 | 3 | ` | 479 | 469 | 763 |
Here, there are at least two occurrences (n4) of an atom in a multiple, non-aromatic bond (n2) located two bonds (n1) away from an atom with at least two heteroatom neighbours (n3). The descriptor sets three keybits (n5), which can be set by other descriptors (n6). The keybits set are 479, 469, and 763 (n7-n9).
A few properties are incompatible, meaning that fewer descriptors are possible than are mathematically allowed. Also, a number of descriptors can be encoded in two different ways (e.g., by reversing n2 and n3) but the algorithm does not encode both options. Thus, 3234 different descriptors encoding occurrence counts of “one or more” are possible. Since occurrence counts up to “999 or more” are allowed, more than 3 million distinct descriptors can be combined into innumerable keysets. How does one select an optimum set of descriptors for use in molecular similarity calculations?
The authors tried to answer this question by adopting the success measure defined in Briem, H.; Lessel, U. Perspect. Drug Discovery Design 2000, 20, 231-244. This is calculated by taking the mean fraction of molecular nearest neighbours which are of the same activity class as the target active compound, and averaging this quantity over all active target compounds. Briem and Lessel’s data set, used by the MDL authors, contains 957 molecules from MDL Drug Data Report (MDDR), 383 of them in active In any one of 5 classes, and 574 inactive in those classes.
Briem and Lessel used Euclidean distances between keys; MDL used Hamming distance. In MDL’s genetic algorithm optimisations they made use of a “training set success measure”, rather than the one based on Briem and Lessel’s data set. The training set consisted of 1700 compounds from MDDR, belonging to 17 active classes, those classes being different from Briem and Lessel’s five.
Both the 166-bit and the 960-bit MDL keysets were used. The latter contains a number of keybits which can be set by more than one descriptor. If all the multiply mapped keybits are removed, a 726-bit keyset is left. Alternatively, each one of the descriptors encoded in the 960-bit keyset can be made to correspond to one unique keybit, yielding a 1387-bit keyset. The author also used a 3234-bit keyset formed by assigning unique keybit to each of the descriptors encoded by the underlying key setting algorithm, assigning an occurrence count to each descriptor of “set one or more times”.
Various approaches to optimisation are possible: steepest descent, information-theory based pruning (surprisal, and surprisal signal to noise ratio S/N), genetic algorithms, and humans using chemical insight. Optimisation is difficult because of the very high dimensional space, because the optimisation surface is very flat, and because there are many local minima.
Taylor showed the following plot for random pruning:
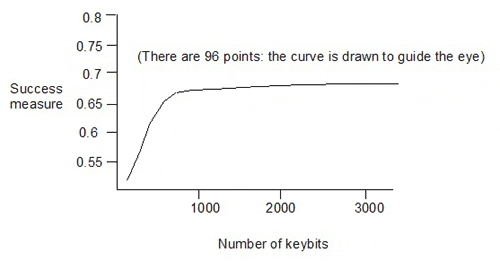
This gives the success measures for 96 keysets, with sizes from 100 to 3200 keybits, generated by eliminating descriptors from the 3234-bit keyset (in which each of the 3234 keybits corresponds to one of 3234 descriptors). The dependence of the success measure on the number of keybits is quite weak above about 1000 keybits in the keyset.
The published results of pruning the keybits from a keyset using surprisal or surprisal S/N ratio as the selection criterion are as follows:
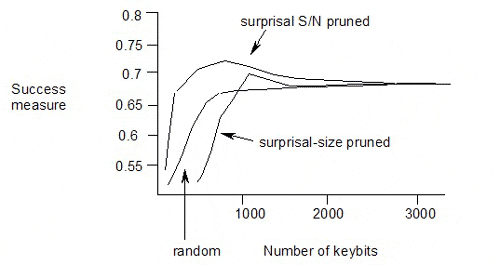
The maximum is at surprisal S/N of 1.9, with 711 keybits used in the keyset, and a success measure of 0.704.
The authors also pruned the 166-, 726-, 960- and 1387-bit keysets and plotted the success measure against the number of keys:
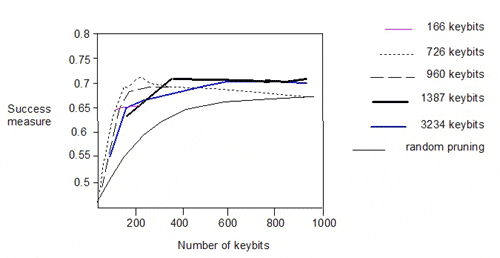
There is significant degradation in performance below 200 keybits. Interestingly, the performance of the 166-bit keyset lies on the curves for the 1387- and 3234-bit keysets. The similar performance of the various pruned keysets does not however, imply commonality in the descriptors encoded in the keysets.
In conclusion, MDL has produced keysets with optimised success measures up to 0.711 compared with success measures of 0.649 and 0.670 for the standard 166- and 960-bit keysets. Surprisal-based selection of keybits was found to be worse than random selection but surprisal S/N base selection outperformed random selection. The great insensitivity of overall performance on both keyset size and identity of the descriptors encoded resulted in great difficulty in optimising the performance using standard mathematical techniques. Genetic algorithm optimisation succeeded in producing keysets with better performance but MDL were not able to locate a single globally optimum keyset.
For hand optimisation, the authors took guidance from the GA and surprisal pruning results, and also considered the definitions of the 166 keys. They considered J. Med. Chem. articles by the Vertex group, showing that current drugs show relatively restricted diversity in terms of ring systems and sidechains. Finally, they gave priority to series descriptors, in which occurrence counts and bond distances varied systematically (e.g., 1, 2, 3 or more occurrences, and 2, 4, and 6 bonds), rather than randomly including particular members. This led to a selection of 324 keys that performed as well or better than the previously examined subsets.
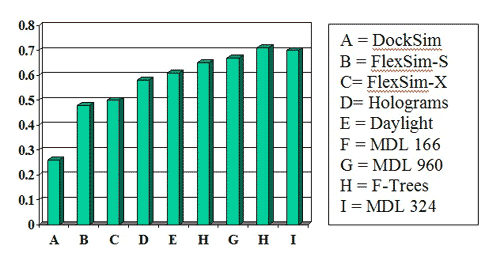
Success measures for the MDL 960-bit and 324-bit keysets were better than nearly all the other methods in two publications by Bemis and Murcko (J. Med. Chem. 1996, 39, 2887 and 1999, 42, 5095):
An experiment in differentiating toxics from drugs was carried out. A database was built by combining MDDR with MDL Toxicity: 141,012 structures were unique to MDDR, 110,138 structures were unique to MDL Toxicity and 1950 structures occurred in both databases. One hundred structures were selected at random. A search was carried out for structures 50% similar to these using weighted Tanimoto similarity. The hitlist size, percentage of hits in MDDR and percentage of hits in MDL Toxicity were recorded.
The authors were interested in the performance of the 324-keybit set compared to the 166- and 960-bit keysets from the standpoint of efficiency and relevance (i.e., does a weighted similarity search bring in a lot of hits, including irrelevant ones, or does it hit a select number of relevant structures). They measured hitlist size and percent MDDR structures of the given hitlist, using a 50% similarity search against 100 random query structures from MDDR, and using three types of weighting The resulting hitlist sizes for MDDR were as follows:
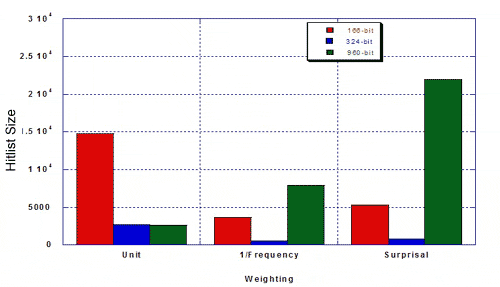
The percentage of true hits for MDDR was as follows.
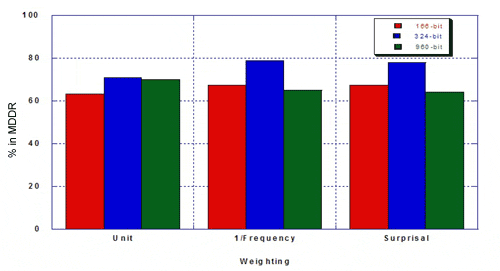
Taylor concluded that the re-optimised keyset outperformed existing 166- and 960-bit keysets: hitlists were more focused and there was a higher percentage of the desired class. Weighting improves the optimised keyset more than it improves existing 160- and 960-bit keysets. Key performance can be improved by re-optimising keysets. Key performance is not substantially improved for MDL keysets longer than about 500 bits. The hand-optimised keyset outperforms 166- and 960-bit keysets in differentiating structures in MDDR from structures in MDL Toxicity.
MDL keys can be accessed as an option in MDL ISIS/Host and the MDL Relational Chemistry Server databases but a structure must be registered. They can also be accessed through the MDL Chemistry Rules interface which takes an MDL molfile as input. In that case, registration into a database is not required. A 320- keybit set has now replaced the 324 keybit set discussed in this presentation. It is not “official”, results are not guaranteed, and the technology is under continuous refinement, but with these caveats, Taylor is willing supply a current version to interested parties.